
聚集诱导发光(Aggregation Induced Emission, AIE)材料由于其独特的发光性质成为了近些年来材料研究领域明星材料,而对其机理和相关理论设计的也是重要的课题。然而由于分子聚集态系统过于复杂,因此对该体系的激发态性质进行全尺寸的第一性原理计算对于现有的量子力学方法几乎是不可能完成的任务,因此研究者们不得不使用一些近似模型给予研究。目前,对AIE材料的第一性原理广泛采用量子力学经典力学杂化(QM/MM)模型。在该近似模型下,中心激发分子用高精度量子力学方法,而周围的环境分子用经典的分子力学考虑,中心分子与环境分子的相互作用一般用静电耦合作用考虑。虽然QM/MM方法的计算为聚集态发光体系的理论研究做出了巨大贡献,但是由于其模型的局限性,对聚集态体系的量子效应的考虑并不完全,所以对分子聚集态体系的光谱的高精度预测仍然非常困难。因此,目前利用第一性原理计算对AIE进行精确理论预测并指导设计是一件非常具有挑战性的课题。
另一方面,近年来,基于分而治之理念的分子片量子力学方法(Molecular fragmentation method)得到了迅速发展。分子片方法的基本原理是利用体系的电子态的局域性,将大尺寸体系分割为众多小尺寸片段,分别进行量子计算,然后将其线性组合,从而获得整个体系的性质,从而使得对大尺寸体系的全量子计算变为可能。分子片方法在预测大分子体系的能量、结构、振动光谱等基态物理化学性质等方面表现出和全量子力学方法高度一致的精确性,并具有接近线性标度的计算量。这些方法的发展启发我们,是否可以用分子片方法对AIE体系进行全量子力学的激发态计算,从而获得更加准确的光谱预测。
基于这个想法,北京化工大学的李晖课题组与华东师范大学的何晓课题组合作,利用他们发展的静电嵌入的扩展化的分子碎片方法(EE-GMF)结合分子动力学模拟和迭代几何优化等的策略,对一类典型的AIE分子聚集体(di(p-methoxylphenyl) dibenzofulvene,FTPE)进行了吸收光谱和发射光谱的全量子计算。结果发现,在该系统中,当同时考虑了体系所有的近程两体间的量子相互作用和远程静电嵌套之后,分子片方法预测的理论光谱与实验光谱的符合程度要远好于单分子计算和QM/MM模型。尤其是分子片全量子力学计算得到的三种聚集形态(两种分子晶体和一种无定型型态)的吸收光谱(TD-B3LYP/CCPVDZ水平)与实验值吸收谱相差小于5纳米,发射光谱相差小于20纳米。全量子计算得到的光谱振子强度也能定性吻合实验测量的几种形态分子聚集体的荧光量子产率。此外,通过基态和激发态的几何结构分析,作者发现传统的基于单分子运动的发光机制,例如分子内旋转旋转受限(restriction of intramolecular rotation,RIR),只能部分解释AIE行为,而近程轨道耦合的改变对该体系的奇特光谱行为有更为重要的贡献。
由此可见,新近发展的分子分块量子力学方法可以成为研究聚集态诱导发光的有力工具,有希望用于理论指导和设计新的聚集态发光分子材料。此外,由于静电嵌入型分子片方法是一种可以扩展的开放型计算平台,通过改变分块的策略和量子计算水平,原则上可以在现有计算水平允许的前提下不断提高理论预测精度。因此,随着高性能计算机的飞速发展,该方法将会成为凝聚态体系激发态行为研究的越来越重要的手段。该工作实验部分的数据和相关讨论来源于北京化工大学顾星桂教授,并以“Quantitative Prediction of Aggregation-Induced Emission: A Full Quantum Mechanical Approach to the Optical Spectra”为题发表在Angew. Chem. Int. Ed, (DOI: 10.1002/anie.202003326) 上,第一作者为北京化工大学张伟博士,通讯作者为北京化工大学李晖教授,顾星桂教授,华东师范大学何晓教授(论文作者:Wei Zhang, Jinfeng Liu, Xinsheng Jin, Xinggui Gu, Xiao Cheng Zeng, Xiao He, Hui Li)。
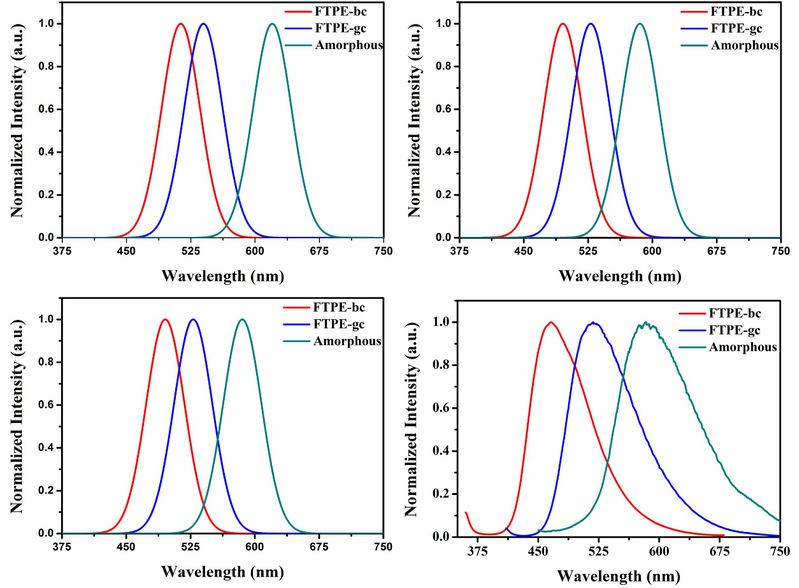
图一、基于单体计算(a)、QM/MM方法(b)和EE-GMF方法(c)计算了FTPE在凝聚相中的发射光谱。(d)实验测量获得的FTPE的发射光谱。(所有的光谱都是用归一化的强度绘制的)

图二(a)对于传统QM/MM方法,只有中心分子(紫色)包含在QM区域内,其他所有分子都用电荷来描述。(b)对于理想的QM/MM方法,中心分子和周围分子(距激发中心6.0 A以内,蓝色)都包含在QM区域内,而晶体环境中的所有其他分子都用电荷来描述。理想的 QM/MM模型的计算成本超出了传统QM计算的极限。(c)对于EE-GMF方法,以中心片段为激发中心,采用多体展开法处理附近分子与激发中心之间的QM相互作用(二体分子间的量子力学相互作用)。

